Published: NoSpherA2
Our paper about refinement using non-spherical form factors has just been published in the RSC’s flagship journal Chemical Science – take a look here:  Paper
Paper
NoSpherA2 – the part of Olex2 that generates the non-spherical form factors – is now also fully functioning and present in all versions of Olex2-1.3.
The section below is a very basic summary of what this is about; this is a kind of ‘press release’ to the full paper, where you
Introduction to refinement using non-sphrical form factors
Knowledge of the exact location of the atoms in a molecule or ionic material is an essential prerequisite to the understanding of chemical reactivity and properties.
X-ray crystallography has served very well for over half a century now in providing just this information, and the presence of a crystal structure is often – rightly – seen as absolute proof of the nature of a material.
At the very heart of X-ray crystallography lies the interaction of X-rays with the electrons in the crystalline material, which gives rise to the diffraction we can measure during the experiment.
There is a simple relationship: more electrons give rise to more diffraction – and this is where we hit a snag: the positions of hydrogen atoms, so central in the understanding of the reactivity and properties of any material, can be very hard to locate in their true position. The single electron inevitably takes part in bonding and thereby diminishes its ability to interact with X-rays even further. If a hydrogen atom is located close to a very heavy element – a metal, for example – then it may be impossible to infer its true location from X-ray diffraction altogether.
This problem is so prevalent, that the location of almost all hydrogen atoms in the Cambridge Structural Database has not actually been determined as such, but these atoms have simply been added to structures in their calculated geometrically sensible positions – and while this is fully understood by most crystallographers, this fact is rarely discussed or in the chemical community.
And yet, the coordinates of X-ray structures are very often and in good faith used as the starting point for further discussions and quantum mechanical property calculations and the results are then taken at face value.
But without reliable knowledge of the true positions of all atoms in a structure, such results may simply be wrong.
Here we introduce a way to obtain true hydrogen atom positions with nothing more than routine X-ray diffraction data and standard desktop computing facilities – using the free and open-source software described in this paper.
The concept is simple and not at all new: what we are doing here is what has been proposed right from the beginnings of crystallographic refinement, but it was impossible to implement then and – for lack of computing power – it was necessary to introduce a simplification that has, curiously, remained in place to the day: the spherical atomic form factor, which results when atoms in a molecule are treated as completely independent of each other.
The interaction of X-rays with electrons in a crystalline material gives rise to measurable diffraction – and the necessary simplification using spherical atomic form factors didn’t matter at the time because the quality of the measurement was also very rough. But this has now changed because the quality of these measurements has increased enormously since the early days.
For there are two sides to coin of structure determination: On one side there is the experimental determination of intensities, and on the other side is the (rather involved) process of ‘mimicking’ the observed diffraction through building a model of the entity that gives rise to the diffraction and ‘refining’ this model to the best of our ability to get the closest match with the observed data – a match that is typically measured as a statistical R factor.
During this process, we take the variables of our model (x,y,z, ADPs) and then must make use of some sort of ‘form factor’ – an entity that describes how a particular atom interacts with X-rays) and – in order to calculate that atom’s contribution to the overall diffraction. If we do this for all atoms, we should be able to get close to the measured values.
If the data were perfect (which they never really are, but they’ve been getting really good due to decades of careful diffractometer development), we will still never reach a perfect match, simply because we are still all using an outdated – and now entirely unnecessary – simplification: the spherical form factor (or, rather the fact that we treat atoms as if they were merely non-interacting spheres): The very way we conduct crystallography assumes that there is no chemical bonding!
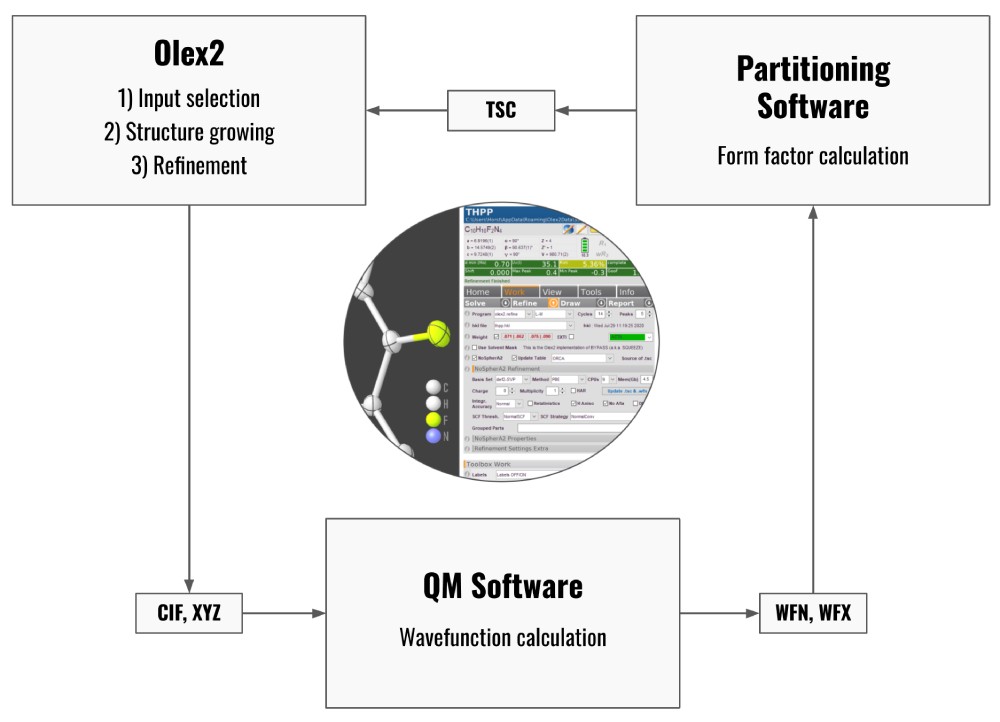
There are various approaches to addressing this issue – and here we concentrate on those that replace the spherical form factors with appropriate non-spherical form factors. For this to work, we need two components: 1) the ability to obtain these form factors and 2) the ability to use them in a refinement program.
The popular small-molecule program Olex2 has a built-in structure refinement engine (called olex2.refine), which is a fully-fledged refinement program that can handle almost all common structure refinements. And now, it can also base its refinement work on non-spherical form factors while otherwise working exactly the same as when using old-fashioned spherical form factors.
So, where do these non-spherical form factors come from? As far as olex2.refine is concerned: it doesn’t really matter – as long as these are provided in a format we have specified previously.
There are two principal routes to these form factors: One can either make use of databases and use the form factors for atoms in similar chemical environments (and which have been either determined experimentally or calculated previously), or one can calculate a unique, custom-made form factor for each atom the exact environment it is present in the structure. This is the route we are describing in this work. It turns out that this can be done using standard laptop or desktop computers in a matter of minutes (for small structures, like sucrose) up to a few hours for all systems that can be considered to be suitable for this analysis in the first place.
What this means is: everyone with access to standard diffraction equipment and standard-quality crystals can get correct and accurate positions of all atoms in a structure, including all hydrogen atoms.