Molecular Orbitals
The calculation of molecular orbitals by their very definition is always delocalized. Canonical Orbitals are always spread-out over the molecule.
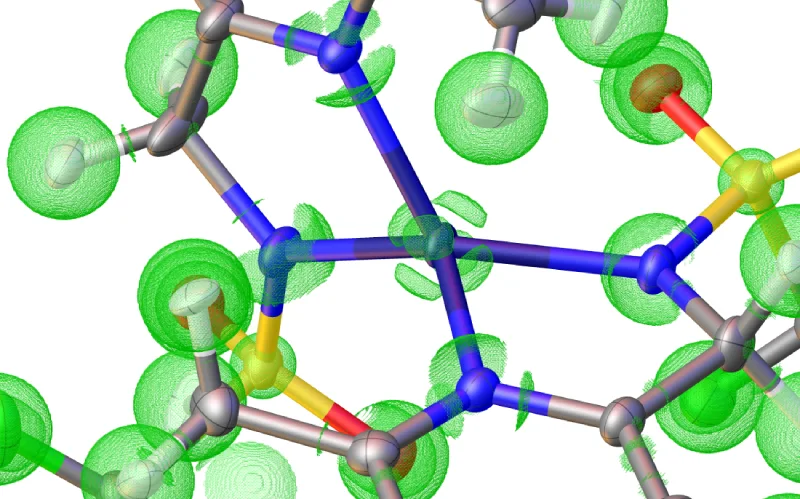
A nice way to visualize bonding without the use of fuzzy orbital localization schemes is the use of spatial electron localization descriptors. One example is the ELI (or the more outdated version, the ELF) or the Laplacian of the electron density.
Here you can usually see bonds, lone-pairs, and valence deformations quite nicely. They are possible to compute from the wavefunction used in NoSpherA2. If you want to do that please have a look at the “NoSpherA2 Properties” Tab, where you can compute and visualize them.
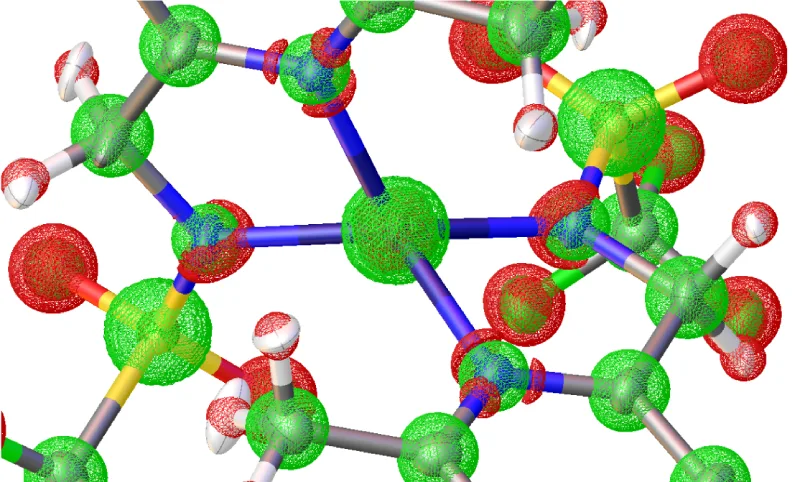
If you want to look at localized orbitals you can have a look at techniques like NBO or NLMO, Boys orbitals or other localization techniques. These attempt to minimize the spatial extension of orbitals by applying different schemes to make linear combinations. NBO tries to map them to the very old concept of Lewis-structures, Boys orbitals are simply minimized in spatial extent and therefore usually visually more appealing than canonical orbitals. This all comes at a cost that they are not direct solutions to the Schrödinger equation anymore.