NoSpherA2 Refinement
This dropdown provides all the settings required for the calculation of the .tsc file.
For HAR the definition of methods for both the QM-Software, as well as refinement settings need to be done.
QM Software Options
The following options control the input for the QM-Software and define the level of sophistication of the wavefunction calculation.
Basis (Set)
This specifies the basis set for the calculation of the theoretical density and wavefunction. The default basis set is def2-SVP.
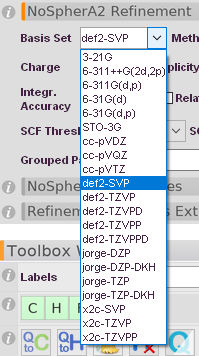
Selection for Basis Set
STO-3G is recommended only for test purposes.
The def2- and cc- series of basis sets is only defined for atoms up to Kr. Afterword ECPs (Effective core potentials) would be needed to use these basis sets, which are by the nature of the method not compatible. So these Basis Sets will not work with any element that has Z higehr than Kr.
The jorge and x2c-basis sets are usefull, as they are defined up to Rn without any ECPs. -DKH and x2c basis sets require relativistic methods (see below).
It is highly recommended to run basic first iterations using single zeta valence basis sets and finish the structure with a higher basis to reduce computational time and sources of errors in calculation setup.
Method
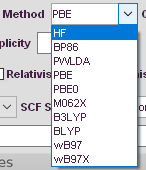
Selection Method
The Method affects the calculation time and accuracy directly. A “cheap” method might not describe the molecular wavefunction correctly, while a extremely exagerated functional might not improve the result of the refinement compared to a more simple one.
The default method used for the calculation of the theoretical MOs/Density is B3LYP. It may be superior in some cases, but GGA functionals like PBE are much faster and less ressource costly. Hartree-Fock is usually not recommended, since DFT can describe electron correlation much better while still doing a decent job with electron exchange. A variety of functionals are available for convenient use. If you need more functionals contact Florian Kleemiss.
Please note that the selection of available methods is limited based on the software used and how extensively the interface was implemented. If you really want to try a method feel free to contact the Olex2 Team for inclusion of your favourite DFT functional.
CPUs
The number of CPUs to use during the waverfunction calculation and partitioning. It is usually a good idea to not use all of them – unless you don’t want to use the computer for anything else while it runs NoSpherA2!
If you are on Windows, and the drop-down box shows only 1 CPU, then you need to install the MS-MPI extensions on your system. You can download the necessary installer from here.
Olex2 will be locked during the calculation to prevent inconsitencies of files. Mostly copying files etc needs overhead CPU capacity, so please leave 1 CPU for these kinds of things. Also note: It is not recommended to run jobs in a Dropbox Folder… They tend to break.
Memory
The maximum memory that NoSpherA2 is allowed to use in GB. This must not exceed the capabilities of the computer. If ORCA/pySCF are used the memory needs per core are calcualted automatically, this input is the total memory.
ORCA requires a certain amount of RAM per core, while pySCF can work in so-called shared memory mode. This reduces Memory requirements drastically when using multiple CPUs. If you encounter problems with ORCA and insufficient Memory it might be worth a try in pySCF.
Charge
The charge of the molecule used for the wavefunction calculation. This must be properly assigned. If PARTs are present the charge applies to all PARTs.
Multiplicity
The multiplicity (2 * S + 1) of the wavefunction, where S is the total spin angular momentum of the configuration. E.g. a closed shell wavefunction in it’s ground-state usually has multiplicity 1, one unpaired electron has multiplicity 2. Must be positive above 0.
Integr.(ation) Accuracy
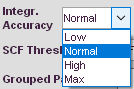
Integration accuracy
Select which accuracy level is requested for the integration of electron density. This affects time needed for calcualtion of .wfn and .tsc files. Normal should be sufficient in most cases. Extreme is mainly for benchmark and test purposes. If you have high symmetry or very heavy elements and high resolution data it might improve results to switch to a higher value. Low can be used, if the number of electrons after integration is sill correct. Mostly fine for pure organic molecules.
Please check in the log files, whether this is the case!
Use Relativistics
Use DKH2 relativistic Hamiltonian. This should only be used with -DKH or x2c-family basis sets. But for them it is highly recommended. In the case of pySCF the x2c relativistic method is used. This is, however, still in testing.
Tonto specific Options

Tonto-specific options available
These Options are only applicable to Tonto as QM-Software.
Cluster Radius (For Tonto)
If Tonto is used for a wavefunction calculation a clsuter of explicit charges calcualted in a self consistent procedure is used to mimic the crystal field for the wavefunction calculation. This option defines the radius until no further charges are included.
Complete Cluster (For Tonto)
In case of a molecular structure this switch will try to complete the molecules for the charges calcualtion bsaed on distances of atoms. If the refined structrue is a network compound where no molecular boundary can easily be found (e.g. there is no way to grow the structure without having dangling bonds) this procedure will fail in a sense, that the computer will run out of memory. Therefore this option is highly recommended for molecular crystals but crucial to be deactivated for network compounds.
DIIS Conv. (For Tonto)
This option defines the convergence criterion the wavefunction calculation needs to achieve in order to be considered converged. A lower value will finish faster but drastically increases the chance for unreasonable wavefunctions, especially in complicated calculations.
ORCA specific Options
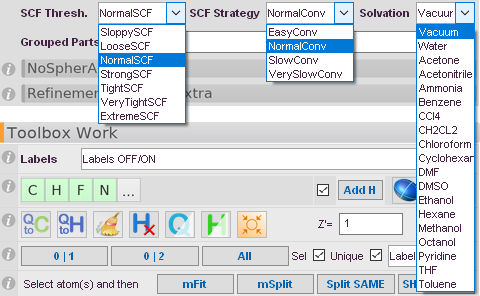
ORCA-specific options available
These Options are only applicable to ORCA as QM-Software.
SCF Conv. Thresh.
This option allows you to adjust the convergence criteria for your SCF in ORCA. NormalSCF for default calculations, Tight or very Tight for very precise high level calculations. Extreme is basically the numerical limit of the computer and strongly discouraged, as practically unreachable for big (>3 atoms) systems.
SCF Conv. Strategy
Selects which mechanism to use for the SCF to converge to the minmum. Refers to the stepsize in applying calculated gradients fo the wavefunction.
Solvation Model
You can embed your wavefunction in an implicit solvation model to mimic polarization effects of the crystal environment. A series of pre-tabulated solvents is accessible.
pySCF specific Options
pySCF-specific options available
These Options are only applicable to pySCF as QM-Software.
Damping
A factor to control the convergence of the wavefunction calculation. If it is selected too high things run away, too low and the calculation takes unnecessarily long. For metal complexes a higher value is recommended while organics usually converge fast with 0.6.
Solvation Model
You can embed your wavefunction in an implicit solvation model to mimic polarization effects of the crystal environment. A series of pre-tabulated solvents is accessible.
Refinement Options
Refinement options available
These options apply to the refinement setup, e.g. handling of hydrogen atoms, release of constraints or handling of disorder.
HAR // Iterative
Repeat calculations until final convergence is achieved over a full cycle of .tsc and Least Squares refinement. Criteria as per definiton of HAR in Tonto: <0.001 shift/ESD. This will need much more time than a single step. But in case you use ORCA or pySCF the last wavefunction is used as initial guess, which will save a lot of time on the consecutive steps.
Max Cycles
This defines a criteria for the abortion of HAR, if convergence is not achieved. Maybe restraints or better parameters (basis, method, grid accuracy etc.), resolution cutoffs or other improvements to your model might help with convergence.
Update .tsc & .wfn
This button will only generate a new .tsc and .wfn file, without running a least squares refinement. This is usefull after a converged model was found to get the most up-to-date wavefunction for property plotting or to test things before submitting a bigger calculation.
H Aniso
Make hydrogen atoms anisotrpically. Make sure they are not restricted by AFIX commands to obtain reasonable results. If not selected no changes will be applied.
No Afix
Remove any AFIX constraints from the current model. Use with caution, but highly usefull for starting from IAM.
DISP
Add DISP Instructuion to your .ins file based on Sasaki table as implemented in Olex2. DISP instructions are needed for correct maps and refinements in case of non metal-based wavelengths (i.e. synchrotron radiation).
Grouped Parts (For disordered structures)
This is an advanced feature still being tested.
Since there might be different regions in your molecules containing disorder modelling you need to specify which disorders form a group for the calculation of Wavefunctions. A Group in this sense refers to PARTs, that resemble the same functional space. E.g. a disorder of a sodium atom with other singly charged ions around this position form a group (Let’s say PARTs 1, 2 and 3 are PARTs describing three possibilities of this disorder), where these different PARTs are not interacting within the same molecular input structure, while at a second position there is a carboxylate in a disordered state, where one of the PARTs interacts with this disordered ion (PART 4), while the second PART (Nr. 5 in this case) does not.
For the given example a reasonable grouping would be: 1-3;4,5
This would mean 1-3 are not to be interacting with each other, but each of them both with 4 and 5, respectively, leading to calcualtions:
PART 1 & 4
PART 1 & 5
PART 2 & 4
PART 2 & 5
PART 3 & 4
PART 3 & 5
Groups are given by syntax:
1-4,9,10;5-7;8
(Group1=[1,2,3,4,9,10] Group2=[5,6,7] Group3=[8])
It is easily understandible that having interactions between PART 1 and 2 in this example would be highly unphysical, which is why definition of disorder groups is crucial for occurances of disorder at more than one position.
IMPORTANT: Superposition of two atoms with partial occupation freely refined without assignment to PARTs will lead to SEVERE problems during the wavefunction calculation, as there will be two atoms at 0 separation. Most likely the SCF code will give up, but will NEVER give reasonable results!